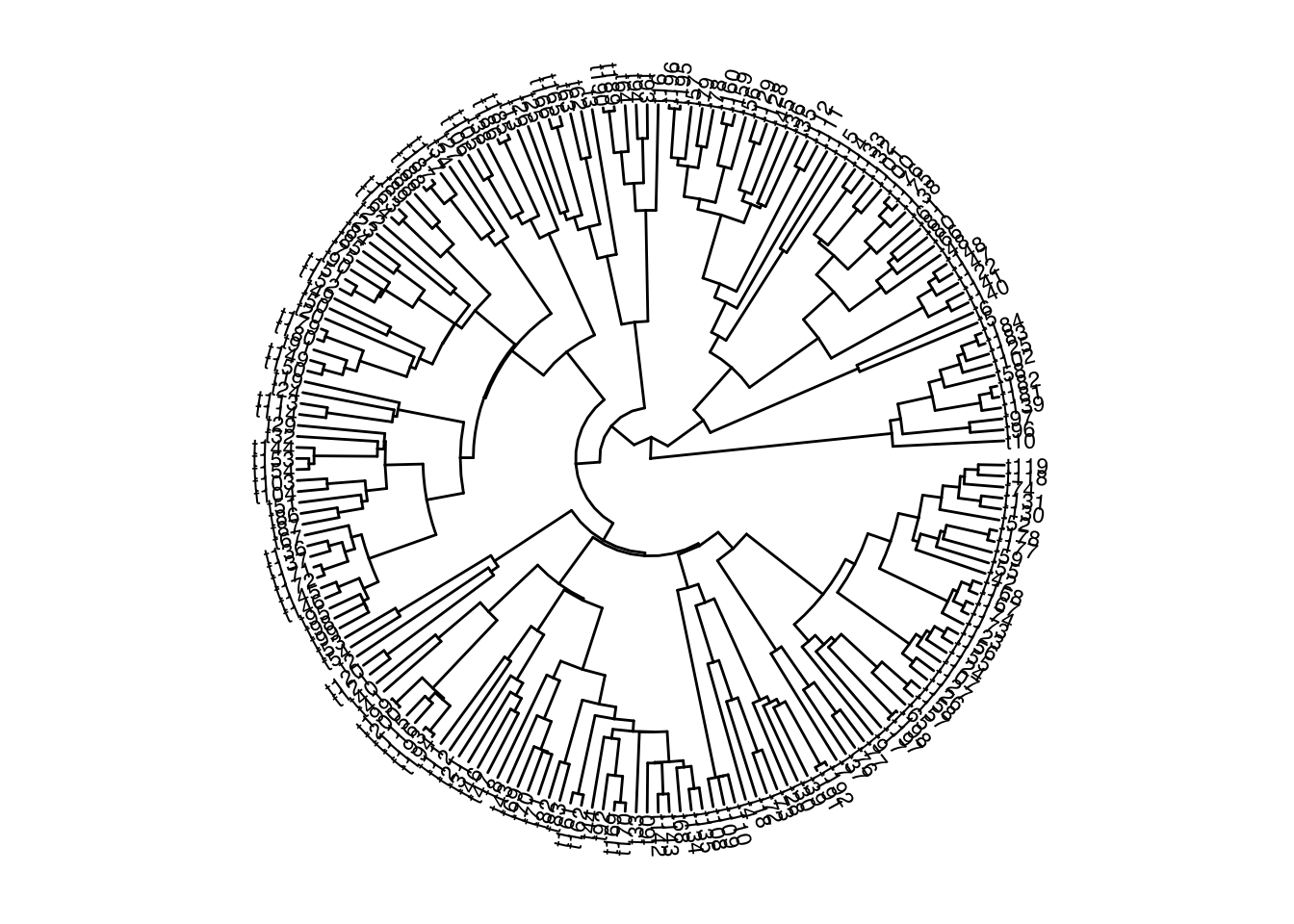
In a current research project, I have a single large phylogeny and tip data for a large number of traits. However, not all tips have data available for all traits. I want to visualise this patchy data coverage by overlaying the sub phylogeny of tips with data for a given trait, over the full phylogeny. This would also be useful to visualise which tips haven been dropped from a phylogeny. However, as far as I can tell, such a method is not directly implemented in any of the big phylo R packages. Here is my version:
First we simualte a tree using phytools::pbtree(). To simulate the patchy trait coverage, we draw (in this example) 4 random samples of the tree’s tips. These are the tips with data for each of the 4 simulated traits.
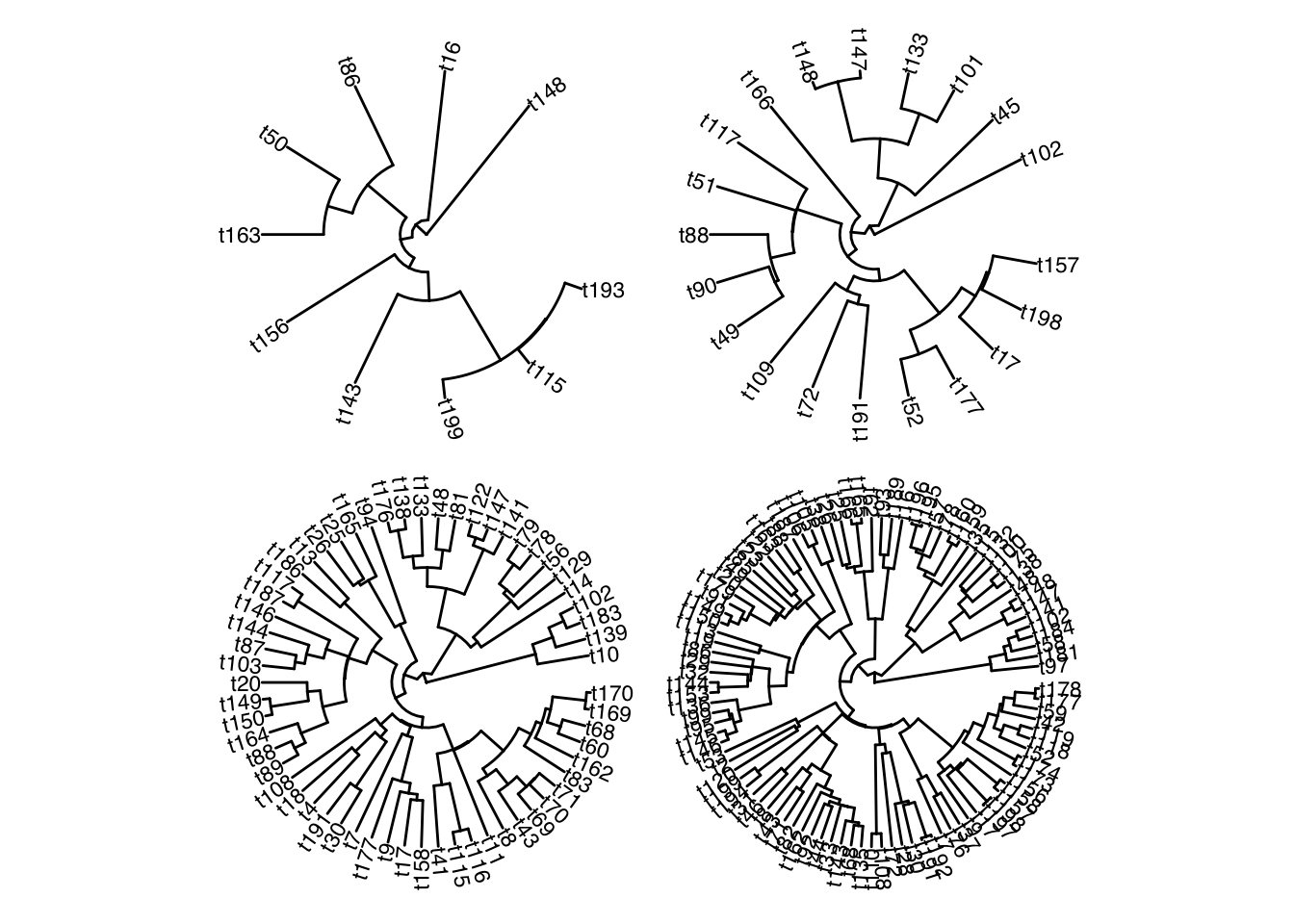
I find it quite hard to tell which tips or clades have been dropped by looking at these trees, even with the tip labels. So instead, let’s plot these trees over the original tree, retaining the original topology. Here’s my attempt at that:
I think the output makes it easy to quickly visually assess what clades have data for a given trait, and which traits are particularly data poor/rich. I understand that this plot shows the same data as a phylogeny and heatmap style plot as shown in the treedata book or the phytools blog, but for my particular purposes this style of plot has additional value, due to the large number of traits.
That’s all!
Source Code
---title: "Plotting the branches of a trimmed phylogeny on the original tree topology"author: "Iain R. Moodie"date: 2024-10-15categories: [R, methods, phylogeny]draft: falseformat: html: code-fold: true code-tools: true code-overflow: wrapexecute: echo: true warning: false eval: true---In a current research project, I have a single large phylogeny and tip data for a large number of traits. However, not all tips have data available for all traits. I want to visualise this patchy data coverage by overlaying the sub phylogeny of tips with data for a given trait, over the full phylogeny. This would also be useful to visualise which tips haven been dropped from a phylogeny. However, as far as I can tell, such a method is not directly implemented in any of the big phylo `R` packages. Here is my version:First we simualte a tree using `phytools::pbtree()`. To simulate the patchy trait coverage, we draw (in this example) 4 random samples of the tree's tips. These are the tips with data for each of the 4 simulated traits.```{r}#| output: false#| echo: falselapply(c("ape", "phytools", "phangorn", "tidytree", "ggplot2", "patchwork", "ggtree", "ggplot2"), function(pkg) {if (!requireNamespace(pkg, quietly =TRUE)) {install.packages(pkg) }})``````{r}set.seed(666)full_tree <- phytools::pbtree(n =200)sample_tip_labels <-function(tree, num_samples =4) { tip_labels <- tree$tip.label sample_sizes <-length(full_tree$tip.label) *c(0.05, 0.1, 0.3, 0.5) samples <-lapply(sample_sizes, function(size) sample(tip_labels, size))return(samples)}tips_with_data <-sample_tip_labels(full_tree)```Here's the phylogeny in full:```{r}ggtree::ggtree(full_tree, layout ="circular") + ggtree::geom_tiplab(size =3)```And here is the subtree for each of the traits, produced by dropping tips/branches for which we lack data for a given trait:```{r}lapply( tips_with_data,function(tips) ggtree::ggtree( ape::keep.tip( full_tree, tips ),layout ="circular" ) + ggtree::geom_tiplab(size =3)) |> patchwork::wrap_plots()```I find it quite hard to tell which tips or clades have been dropped by looking at these trees, even with the tip labels. So instead, let's plot these trees over the original tree, retaining the original topology. Here's my attempt at that:```{r}#| classes: preview-imageplot_subtree <-function(tree, tips, layout ="circular") { get_substree_nodes <-function(tree, tips) { mrca <- phangorn::mrca.phylo(tree, tips) nodes <-unique(unlist(lapply(seq_along(tips), FUN =function(i){phangorn::Ancestors(tree, tips[i], "all")} ) ) )return(nodes[nodes>mrca]) } nodes_to_colour <-get_substree_nodes(tree, tips) tree_dat <- tree |> tidytree::as_tibble() |> dplyr::mutate(colour_nodes = dplyr::case_when( node %in% nodes_to_colour ~TRUE, label %in% tips ~TRUE,TRUE~FALSE ) ) |> tidytree::as.treedata()return( ggtree::ggtree(tree_dat, ggplot2::aes(colour = colour_nodes), layout = layout) + ggtree::geom_tippoint(ggplot2::aes(colour = colour_nodes, shape = colour_nodes)) + ggplot2::scale_colour_manual(values =c("grey85", "black")) + ggplot2::scale_shape_manual(values =c(NA, 16)) + ggplot2::theme(legend.position ="none") )}plots_circular <-lapply( tips_with_data,function(tips) plot_subtree(full_tree, tips) )patchwork::wrap_plots(plots_circular)```I think the output makes it easy to quickly visually assess what clades have data for a given trait, and which traits are particularly data poor/rich. I understand that this plot shows the same data as a phylogeny and heatmap style plot as shown in the [treedata book](https://yulab-smu.top/treedata-book/chapter7.html#gheatmap) or the [phytools blog](http://blog.phytools.org/2016/01/new-function-for-phylogenetic-heat-map.html), but for my particular purposes this style of plot has additional value, due to the large number of traits.That's all!